Battery informatics

Impedance-based forecasting of lithium-ion battery performance amid uneven usage
How a battery degrades depends on how it was used in the past. A key challenge in the electric vehicle industry is predicting future battery performance, when different drivers use their cars differently. We tackle this challenge by developing a way to non-invasively probing the internal state of a battery. Coupled with machine learning, this allows us to predict how batteries would degrade given future use profiles.

Identifying degradation patterns of Li-ion batteries from impedance spectroscopy using machine learning
Forecasting the state of health and remaining useful life of Li-ion batteries is an unsolved challenge that limits technologies such as consumer electronics and electric vehicles. We build an accurate battery forecasting system by combining electrochemical impedance spectroscopy (EIS) - a real-time, non-invasive and information-rich measurement that is hitherto underused in battery diagnosis - with Gaussian process machine learning.
Design of inorganic materials

Rapid discovery of stable materials by coordinate-free coarse graining
Predicting the structure of a material given its composition is one of the longstanding challenges in materials science. We address this problem by developing a new way to coarse-grain the structure-composition space of materials. Our approaches leads to rapid and accurate inorganic crystal structure prediction.

Materials informatics reveals unexplored structure space in Cuprate superconductors
We uncover new structure-Tc relationships in Cuprate superconductors using machine learning which guide future nano-engineering efforts. We mine the crystal structure data for high Tc Cuprate superconductors, and develop a materials informatics approach to combine multiple sources of mixed fidelity data.
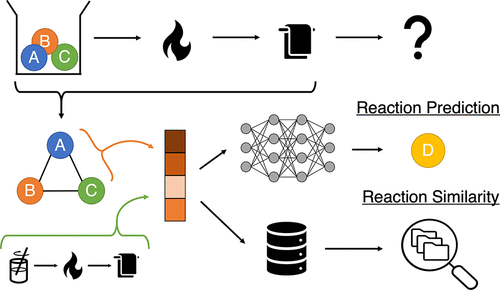
Predicting the outcomes of material syntheses with deep learning
A key bottleneck for material discovery is synthesis. While significant advances have been made in computational material design, synthesis pathways are still often determined through trial and error. We develop a method that predicts the major product of solid-state reactions. Our approach also establishes a quantitative metric for inorganic reaction similarity, allowing the user to explain model predictions and retrieve relevant literature sources.

Predicting materials properties without crystal structure: Deep representation learning from stoichiometry
Machine learning can accelerate materials discovery by accurately predicting materials properties with low computational cost. However, the model inputs remain a key stumbling block: current methods typically use hand-engineered descriptors or use the full crystal structure. We develop a machine learning approach that takes only the stoichiometry as input and automatically learns appropriate and systematically improvable descriptors from data. We show that our method can predict properties such as the critical temperature of superconductors.
Structure and dynamics of electrolytes

Bayesian unsupervised learning reveals hidden structure in concentrated electrolytes
Electrolytes play an important role in a plethora of applications ranging from energy storage to biomaterials. Notwithstanding this, the structure of concentrated electrolytes remains enigmatic. We reframe the problem into the language of computational statistics, and test the null hypothesis that all ions share the same local environment. Our statistical technique reveal distinct local ionic environments; surprisingly, these differences arise in like charge correlations rather than unlike charge attraction. The resulting fraction of particles in non-aggregated environments shows a universal scaling behaviour.

Screening lengths in ionic fluids
The decay of correlations in ionic fluids is a classical problem in soft matter physics that underpins applications ranging from controlling colloidal self-assembly to batteries and supercapacitors. We develop a model that explains the recently observed discontinuous change in the structural force across a thin film of ionic liquid-solvent mixtures as the composition is varied, as well as reframes recent debates in the literature about the screening length in concentrated electrolytes.

Scaling analysis of the screening length in concentrated electrolytes
We developed a scaling theory which shows why the electrostatic correlation length in concentrated electrolytes could be an order of magnitude larger than the ion diameter. We also showed that the electrostatic correlation length is directly related to bulk thermodynamic properties such as the activity coefficient.